“Edison is the go-to for a new investor to get full understanding of the investment case and business. Edison’s distribution reaches investors that we want to connect with directly.”
Join us for investment knowledge across your choice of sectors and equities.
To continue reading, log in or create a free account below.
Close
Close
Close
Create An Account
If you have an account with us, please click the sign in button below.
Redesigning immunity: The next frontier in immuno-oncology
Healthcare
Redesigning immunity: The next frontier in immuno-oncology
Immuno-oncology (IO) has transformed cancer treatment, shifting the paradigm from cytotoxic and targeted therapies to immune-driven disease control (harnessing the body’s own immune system). Immune checkpoint inhibitors (ICIs), CAR-T cell therapies and cancer vaccines have produced durable responses in defined patient populations; however, the first wave of IO is approaching clinical and commercial saturation. ICI response rates remain limited to c 20–40% in solid tumours, while CAR-T therapies, despite curative potential in haematological malignancies, are restricted by high cost, complex manufacturing, immune-related toxicities and lower efficacy in solid tumours. Earlier cancer vaccines also failed to show consistent benefit. These constraints are driving next-generation approaches, focused on platforms that actively programme immune responses, rather than simply releasing inhibitory checkpoints. This report examines key innovation vectors shaping the next phase for this sector.
New-generation ICIs set to break biological ceiling
With PD-1/CTLA-4 franchises such as Merck’s Keytruda (>$30bn in sales in 2025) reaching maturity, incremental value is shifting to next-generation checkpoints that can address adaptive resistance, extend duration of response, expand responder populations and support lifecycle management. Bristol Myers Squibb’s (BMS’s) Opdualag (LAG-3/PD-1 combination) provides the first commercial proof that dual-checkpoint strategies can extend durability and defend PD-1 backbones. We expect assets showing additive efficacy with PD-1/CTLA-4s and favourable safety are most likely to secure partnering and premium valuations.
Cancer vaccines as immune-priming platforms
Next-generation cancer vaccines are transitioning from monotherapy setbacks to high-value immune-priming partners capable of generating high-avidity, tumour-specific responses and converting immunologically cold tumours into ‘hot’ tumours. Moderna/Merck’s mRNA vaccine V940/mRNA-4157 provides the clearest validation in melanoma studies. We see cancer vaccines’ strongest commercial positioning in combination treatments, earlier-stage and minimal residual disease settings, where durable immune memory supports long treatment windows. Platform scalability, precision biomarker strategies and consistent combination benefit across tumour types will be critical to partnering activity and peak-sales realisation.
CAR-T therapies at an inflection point
CAR-T therapies (such as Gilead’s Yescarta and Novartis’s Kymriah) deliver potentially curative outcomes in select haematological cancers, but uptake remains constrained by limited solid-tumour efficacy, manufacturing complexity, high cost and toxicities. Next-wave strategies, including allogeneic platforms, bi-specific/multi-targeting constructs programmes and CRS-mitigation approaches, promise to reduce infrastructure intensity and enable earlier-line use, and will determine CAR-T’s evolution from niche therapy to a broader commercial modality.
Immuno-oncology: The next phase of value creation
Our immune system is naturally built to detect and eliminate abnormal cells while sparing healthy tissue. However, cancer has evolved several ways to evade immune recognition, allowing unchecked tumour growth. While traditional treatments like chemotherapy and radiation can be effective, they are associated with significant side effects. In contrast, IO is a class of cancer therapies that harness, activate or engineer the body’s natural defence mechanisms to recognise and destroy tumour cells, effectively removing the ‘brakes’ that cancer places on the immune system. Since approval, IO has reshaped cancer care, led by ICIs that have become the backbone treatment across multiple solid tumour types. Yet the first ICI wave has also exposed clear constraints: response rates remain limited in many indications, resistance emerges frequently and some tumour types remain stubbornly ‘cold’. In parallel, haematological malignancies remain an untapped frontier where ICIs have delivered more modest outcomes.
The growing challenge is no longer whether immunotherapy works, but how to make it work for the majority of patients, across a broader range of tumours, with acceptable safety, cost and scalability. The next phase of value creation is therefore less about a single breakthrough drug and more about expanding the responder pool and durability of benefit through new checkpoint targets beyond the current PD-(L)1 and CTLA-4 combination strategies that address tumour-resistance mechanisms and new immunotherapy modalities such as therapeutic cancer vaccines and next-generation cell/CAR-T therapies.
Commercially, this evolution should continue to drive partnering: big pharma’s PD-(L)1/CTLA-4 franchises, broad oncology footprints and overhang from the upcoming patent cliff (>$200bn in revenues at risk by 2030) create a natural pull for differentiated add-ons (novel checkpoints, vaccines, intratumoural immunotherapies) and for technologies that reduce toxicity and the healthcare burden in cell therapies.
This report explores the scientific rationale for next-generation IO approaches, showcases leading biotech innovators advancing the field, examines the commercial dynamics driving deal-making and presents the investment case for biotechs at the forefront of this evolution.
Market context: Oncology dominates therapeutic development
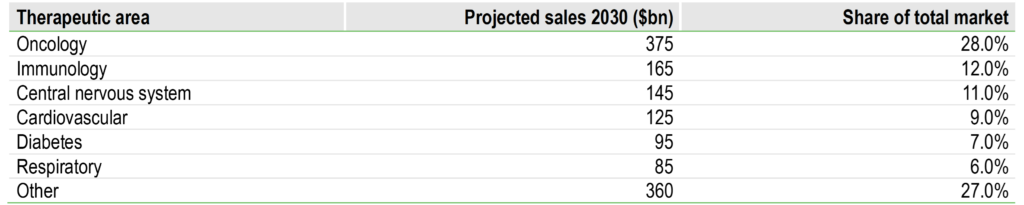
Oncology remains the largest and most innovation-intensive therapeutic area in pharma, reflecting a combination of large addressable patient populations, persistent unmet need and the high economic value of clinically meaningful survival improvements. According to Evaluate Pharma, oncology accounted for 39% of global pharmaceutical R&D spend in 2023, with projected sales of $375bn by 2030, more than twice that of the next largest therapeutic area, immunology, at $165bn (Exhibit 1).
Exhibit 1: Therapeutic area market size projections (2030)
Source: Evaluate Pharma, Edison Investment Research
Within oncology, IO has become a central pillar of R&D and commercial strategy. PD-(L)1 inhibitors have created a large, durable market, but the competitive dynamics have shifted from ‘who has a PD-1’ to ‘who has the best combination ecosystem and differentiated biology’.
This matters for how value is likely to accrue. In the next phase, winners are likely to be those that can: (a) take immunotherapy into harder-to-treat tumours and patient subgroups; (b) move effective approaches earlier in the disease course (adjuvant/maintenance/minimal residual disease); and (c) reduce toxicity and delivery friction so that therapies can be used more widely and earlier.
ICIs: Revolution and limitations
What are ICIs?
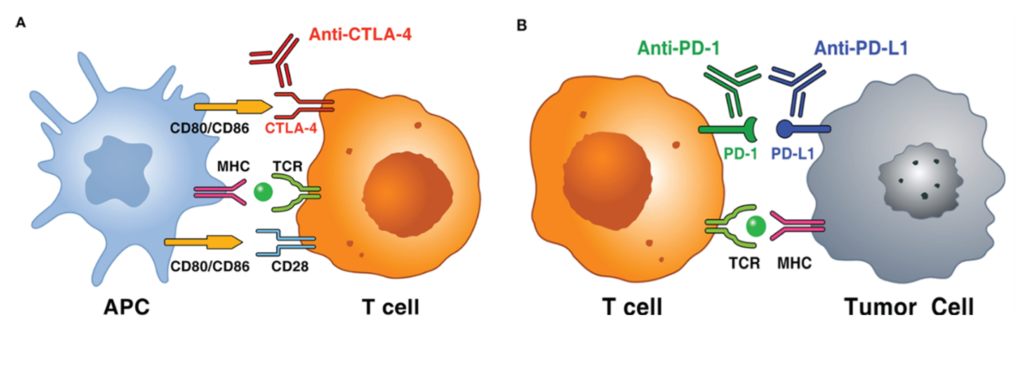
ICIs are immunotherapies or drugs that modulate immune ‘brakes’ that ordinarily prevent excessive immune activation, such as by T cells. Tumours exploit these brakes to avoid immune destruction. By blocking these inhibitory brakes, ICIs restore anti-tumour immune activity, thereby enabling the immune system to recognise and attack cancer cells (Exhibit 2). The two foundational inhibitory pathways to date have been PD-1/PD-L1 (dominant in the tumour microenvironment (TME); affects peripheral effector function) and CTLA-4 (more upstream; affects T-cell priming/activation in lymphoid tissues). By releasing these brakes, ICIs can convert an existing anti-tumour immune response into deep, durable remissions in a subset of patients, an outcome rarely achieved with historical cytotoxic treatments or reliably matched by targeted therapy.
Clinically, the impact has been most visible in immunogenic tumours (with multiple neoantigens; proteins exclusively expressed by cancer cells) and where a pre-existing anti-tumour immune response can be amplified (eg ‘hot’ tumours such as melanoma, non-small cell lung cancer (NSCLC), renal cell carcinoma and classical Hodgkin lymphoma). However, mechanistically this is not a simple ‘on/off’ switch: the effectiveness of ICIs depends on antigen presentation, T-cell trafficking and infiltration, TME suppressive factors and the dynamic expression of multiple immune checkpoints.
Current ICI landscape limited to PD-(L)1s
The current ICI market (Exhibit 3) has been dominated by PD-1 and PD-L1 inhibitors, which have achieved broad approvals, frequent label expansions and deep integration into treatment pathways, with Merck’s Keytruda (generic name: pembrolizumab) leading the pack with $31.7bn in annual sales in 2025 and approval across 20+ indications, with an additional 20+ indications in clinical development. CTLA-4 inhibition (BMS’s Yervoy) remains important in select high-intensity combinations. More recently, LAG-3 joined the approved checkpoint set through BMS’s Opdivo (nivolumab) + relatlimab fixed-dose combination, Opdualag, in melanoma (approved in 2022), the first successful next-generation checkpoint beyond CTLA-4. Opdualag’s approval validates the potential clinical relevance of additional checkpoints, in our view.
Exhibit 3: Approved ICIs
Source: Evaluate Pharma, Edison Investment Research
According to Global Markets Insights, the global ICI market was valued at $66.2bn in 2025, with double-digit growth expected over the next decade, driven by earlier lines/adjuvant settings, new combinations and next-generation mechanisms that can extend benefit beyond today’s responder ceiling (discussed in further detail below).
Limitations of current ICIs
Hitting the biological ceiling: Despite transformative outcomes in some tumour types, only a minority of patients benefit from PD-(L)1 and CTLA-4 blockade, in what is termed a ‘biological ceiling’ issue. Large reviews commonly cite 20–40% of the patients benefiting across broad populations depending on tumour type, biomarker enrichment and treatment regimen. While response rates can vary, the key issue is that a large proportion of patients either do not respond at all or respond transiently before disease progression. This constrains the ceiling of benefit for monotherapy and forces the field toward combination strategies, better patient selection and immune priming approaches.
Resistance mechanisms – innate and acquired resistance: The variability in ICI efficacy is fundamentally driven by tumour immune resistance mechanisms, which can be broadly classified as innate (primary) or acquired (secondary). In primary resistance, tumours lack the biological prerequisites for response, including effective antigen presentation, baseline immune activation and pre-existing T-cell infiltration. In acquired resistance, initial responders subsequently relapse as tumours undergo immune editing, downregulate antigen presentation, activate alternative inhibitory checkpoints or remodel the TME to exclude effector T cells. Together, these adaptive escape processes cap response rates and durability.
Haematological malignancy limitations: While ICIs have transformed outcomes in multiple solid tumours, their performance has often been more variable in haematological cancers. Many haematological malignancies have lower levels of tumour-specific mutations (so the immune system has fewer abnormal targets to recognise) and exist in the bone marrow and lymph node environment, which is naturally immunosuppressive and can physically limit effective T-cell activity. As a result, releasing a single immune brake is often not enough to generate a meaningful anti-tumour response. The key exception is classical Hodgkin lymphoma, where the PD-1 pathway is genetically overactive, making it particularly sensitive to checkpoint blockade.
This has created space for other immunotherapy modalities (including cellular therapies and vaccines) to become central to innovation and value creation in haematology.
Toxicity and immune-related adverse events: ICIs work by activating the immune system, but this broad activation can also cause the immune system to attack normal healthy tissues, leading to immune-related adverse events (irAEs). These side effects can affect multiple organs (most commonly the skin, gut, liver, lungs and hormone-producing glands) and range from mild inflammation to, in rare cases, severe or life-threatening complications. In clinical practice, most irAEs are manageable with early detection and immunosuppressive treatment, but they remain a key factor in treatment selection and in the design of next-generation IO combinations.
TME complexity: ICIs are also less effective in many cancers because of the complex TME (the community of non-cancer cells, blood vessels and signalling molecules that surround the tumour). In some tumours, this environment acts like a protective shield, keeping cancer-killing immune cells out, switching them off, or flooding the area with signals that suppress immune activity. This means that even if an ICI releases the ‘brakes’ on the immune system, the T cells either cannot reach the tumour or cannot function properly once they get there. As a result, the treatment has little effect. Overcoming this barrier is a major focus for next-generation IO, with new strategies aimed at making these tumours more immune-friendly (such as intratumoral therapies, vaccines and myeloid-targeting approaches) so checkpoint inhibitors can work more effectively.
Cold tumours (eg pancreatic, prostate, ovarian, colorectal cancer): ICIs tend to work best when there is already an active immune response against the tumour. In so-called ‘cold’ tumours, this immune activity is largely absent (there are very few cancer-fighting T cells in or around the tumour), and the cancer often does not produce enough abnormal signals to attract them. As a result, giving an ICI is like releasing the brakes on a car that has no engine running – the drug removes the immune block, but there are not enough immune cells present to attack the cancer. In addition, these tumours often create a strongly suppressive environment that keeps immune cells out. To address this, a key focus of current research is on combination approaches that first ‘heat up’ the tumour by bringing immune cells in, and then use ICIs to activate them.
Upcoming newer generation ICI strategies
To transcend current limitations, the field is aggressively exploring novel immune checkpoints and innovative modalities, with the goal of improving response rates, overcoming resistance and broadening benefit to more tumour types and patients.
Expansion into earlier treatment lines: One of the key commercial upsides for ICIs will come from increasing use in the adjuvant, peri-operative and minimal residual disease (MRD) settings, where patient numbers are higher, treatment duration is longer and curative intent supports premium pricing. This is already evident with pembrolizumab in adjuvant melanoma and peri-operative NSCLC (KEYNOTE-671), nivolumab in adjuvant melanoma and resected oesophageal/gastroesophageal junction (GEJ) cancer (CheckMate-577) and atezolizumab in adjuvant NSCLC (IMpower010). With metastatic markets largely penetrated, future growth and blockbuster potential for next-generation IO will depend on demonstrating meaningful benefit in earlier-stage disease.
New-generation checkpoint targets – LAG-3, TIGIT, TIM-3, VISTA: Most solid tumours can express multiple inhibitory checkpoints at the same time, often five to 10 or more across tumour cells, building the biological basis for a ‘multiple brakes’ concept and for the move toward dual- and multi-checkpoint strategies. Beyond PD-(L)1 and CTLA-4, multiple inhibitory pathways have emerged as potential levers to overcome resistance and improve response. However, translating target biology into consistent clinical benefit has been uneven.
Clinical validation: LAG-3 represents the first non-PD-(L)1/CTLA-4 checkpoint to achieve regulatory approval (with BMS’s Opdualag in melanoma), validating the relevance of next-generation checkpoints. While several companies are developing LAG-3-targeting assets, one of the leaders is Immutep, with its programmes targeting the LAG-3/MHC Class II pathway on clinical strategy emphasising immune activation in combination settings. Its LAG-3 inhibitor, LAG525 (ieramilimab), is an ICI out-licensed to Novartis. Interestingly, its lead asset, eftilagimod alpha (efti), also targets the LAG-3 pathway, but is an immune activator rather than a checkpoint inhibitor. Instead of blocking LAG-3, efti activates antigen-presenting cells (eg dendritic cells, monocytes that are central regulators within the immune system bridging adaptive and innate immunity) directly via the MHC Class II pathway, helping them prime and expand cancer-fighting T cells. This upstream mechanism makes it a natural combination partner for PD-1 therapies, as it can generate new tumour-targeting immune cells while PD-1 blockade keeps those cells active. Efti is currently being studied in a Phase III trial (TACTI-004) in combination with the PD-1 inhibitor Keytruda and chemotherapy as a first-line treatment for advanced/metastatic NSCLC.
Setbacks: While LAG-3 has been a success story (notwithstanding the late-stage failure of Merck’s favezelimab in colorectal cancer), other once promising targets such as TIGIT and TIM-3 have had a more challenging journey. The TIGIT class, once seen as a PD-1 successor, has faced a series of late-stage setbacks and is now seen as a higher-risk, selective opportunity. The major clinical disappointments include Roche/Genetech’s tiragolumab (which failed multiple Phase III studies, including SKYSCRAPER-01 and SKYSCRAPER-02, SKYSCRAPER-03 and SKYSCRAPER-14), Merck’s vibostolimab (failure in Phase III KeyVibe-003, KeyVibe-007 and KeyVibe-010 trials) and Novartis/BeiGene’s ociperlimab (failed in Phase III AdvanTIG-302 study). Based on our understanding, AstraZeneca is the only large pharmaceutical company with a significant presence in TIGIT, albeit with its PD-1/TIGIT bispecific rilvegostomig (currently in Phase III).
Emerging targets: One of the most promising emerging checkpoint targets has been VISTA, due to its expression patterns and potential role in immune suppression and resistance mechanism, especially in myeloid-rich tumour environments. VISTA is upregulated in over 30% of cancer tumours and is implicated in resistance to PD-1/PD-L1 therapies, supporting its role as a combination treatment with PD-1s. Australia-based Percheron Therapeutics is a front-runner in this space with its anti-VISTA antibody, HMBD-002. The limited number of active VISTA-related clinical programmes creates an early mover advantage for HMBD-002, the only IgG4 VISTA antibody in development (competitive programmes are all IgG1). Percheron plans to initiate Phase II proof-of-concept trials in Q226.
Combination treatments – two better than one?: With growing proof of the ability of tumours to activate multiple immune ‘brakes’ simultaneously, combination checkpoint strategies are increasingly being evaluated. These aim to widen the responder pool by targeting complementary resistance pathways, deepen responses by enhancing T-cell activation and reduce the likelihood of tumour escape via single-pathway adaptation.
Dual-ICI combinations: This concept is already clinically validated by regimens such as nivolumab (Opdivo; PD-1) + ipilimumab (Yervoy; CTLA-4), durvalumab (Imfinzi; PD-1) + tremelimumab (Imjudo; CTLA-4) and nivolumab + relatlimab (PD-1 + LAG-3). Several other checkpoint combination treatments are under development with next-generation ICIs and PD-1 as backbone, with big pharma increasingly using these regimens as a label-expansion and lifecycle-defence tool.
ICI-targeted therapy combinations: These represent a pragmatic step beyond dual-ICI regimens. By modulating tumour biology, via increased tumour antigenicity and immunogenic cell death (eg, BRAF/MEK, PARP), vascular normalisation and improved immune infiltration/trafficking (eg, VEGF inhibition), or relief of immune exclusion/suppression (eg, CDK4/6, TGF-β pathway targeting), these approaches may increase the likelihood of response to PD-1/PD-L1 blockade in selected settings. Clinically validated ICI-TKI combinations in renal cell carcinoma (such as pembrolizumab + axitinib) and hepatocellular carcinoma (atezolizumab + bevacizumab) provide proof of concept.
ICI-antibody-drug conjugates (ADCs) combinations: This is a rapidly evolving oncology strategy that combines the targeted cytotoxicity of ADCs with the immune-reinvigorating activity of ICIs. ADC-mediated tumour cell killing can promote immunogenic cell death, enhance antigen presentation and increase T-cell infiltration, thereby potentially sensitising tumours to PD-1/PD-L1 blockade. The clinical proof of concept came in 2023 with the approval of Astellas/Pfizer’s enfortumab vedotin (Padcev) plus pembrolizumab (Keytruda) for previously untreated advanced urothelial carcinoma, which has rapidly become the first-line standard of care in this setting.
One thing is clear from the above – to win adoption, combinations need either a clear efficacy step-up (eg overall survival benefit) or a tolerability/convenience advantage that enables broader eligible populations.
Bispecific and multi-specific agents: Bispecific and multi-specific agents attempt to integrate the aforementioned combination logic into a single molecule, potentially improving pharmacology and co-targeting engagement, convenience of administration and intellectual property differentiation. These agents can be designed to simultaneously block multiple inhibitory pathways, bring immune effector cells into proximity with tumours, or combine inhibitory checkpoint blockade with immune stimulation. Early clinical and regulatory validation is emerging, with PD-1 × CTLA-4 bispecifics such as Akeso’s cadonilimab approved in China in first-line treatment in gastric/GEJ cancer in combination with chemotherapy. PD-1 × LAG-3 bispecifics represent a mechanistically validated optionality following Opdualag, with assets such as EpimAb/BeiGene’s EMB-02, aiming to co-target the commonly co-expressed pathways and show activity in PD-1–refractory disease. However, challenges remain, as evidenced by the clinical discontinuations of MacroGenics’s tebotelimab and Roche’s tobemstomig, underscoring execution risk. PD-1 × TIGIT formats, led by AstraZeneca’s rilvegostomig, are higher risk but offer potential upside if bispecific architecture can outperform prior TIGIT monoclonal antibodies. The challenge will be to demonstrate meaningful differentiation, efficacy or tolerability versus simply co-administering two agents, while maintaining manufacturability and safety.
More recently business development activity in ICI bispecifics has increasingly converged on the PD-(L)1 × VEGF axis, which has emerged as one of the most competitive post-PD-1 backbone strategies. VEGF (vascular endothelial growth factor) is a key signalling protein that tumours use to stimulate the formation of new blood vessels (angiogenesis). VEGF-targeted therapies disrupt this process, effectively starving the tumour and normalising its abnormal vasculature. By simultaneously blocking immune checkpoints and normalising tumour vasculature, these agents may help address two of the key limitations of first-generation PD-1 therapy: poor T-cell infiltration and an immunosuppressive microenvironment. A series of large transactions, including multi-billion-dollar collaborations, the largest being the $11.1bn deal between BMS and BioNTech (for pumitamig) in June 2025, highlights big pharma’s urgency to secure next-generation platforms that combine immune activation with TME normalisation. Exhibit 4 lists the major ICI bispecific licensing deals since 2022, the largest volume and value of which have been in PD-1 x VEGF bispecifics.
Value in the ICI space is shifting from new PD-1 monotherapies to combination-enabling assets that expand responder populations, overcome resistance and move treatment into earlier lines. The biological ceiling creates strong partnering demand for next-generation checkpoints, TME modulators and immune-priming approaches. Bispecific deal activity highlights big pharma’s urgency to secure differentiated, franchise-extending platforms. Therapies that demonstrate additive efficacy with manageable toxicity and clear biomarker strategy are most likely to achieve premium valuations and early monetisation.
Cancer vaccines: From disappointment to resurgence
Unlike traditional preventative vaccines, therapeutic cancer vaccines are designed to treat patients who already have cancer by training the immune system to recognise and attack tumour-specific targets, mainly by activating cancer-killing T cells. This is more challenging because cancer comes from the body’s own cells and often hides from the immune system, but the same fact also makes these vaccines a strong partner for checkpoint inhibitors. The vaccine helps create more tumour-fighting immune cells, and the checkpoint drug keeps them active. Earlier-generation cancer vaccines delivered only modest clinical benefit and struggled commercially due to weak immunogenicity, suboptimal antigen selection and deployment in late-stage disease, where immune dysfunction is profound.
However, recent randomised clinical data provide the first clear evidence that newer-generation vaccines can significantly reduce recurrence risk when used in the early or minimal residual disease setting and/or in combination with PD-1 inhibitors. This represents a fundamental shift in how the modality is positioned: not as a monotherapy for metastatic disease, but as an immune-priming platform capable of inducing durable, tumour-specific T-cell responses.
Historical clinical development and limited early impact
First-generation cancer vaccines were based on several technological approaches, including peptide vaccines, whole-cell tumour vaccines and autologous dendritic cell therapies. While these strategies demonstrated proof of principle that immune responses could be generated against cancer, they failed to produce consistent and clinically meaningful tumour regression.
The approval of Dendreon Corp’s sipuleucel-T (Provenge) in 2010 for metastatic castration-resistant prostate cancer represented a landmark for the field, as it showed an overall survival benefit in a randomised trial. However, the absence of objective tumour responses, the complexity of autologous manufacturing, high cost of goods and logistical challenges limited physician adoption and ultimately constrained commercial success.
More broadly, these early programmes suffered from a lack of biomarker-driven patient selection (such as MSI-H/dMMR), poor antigen immunogenicity due to immune tolerance and clinical trial designs that evaluated vaccines in heavily pre-treated metastatic patients with advanced immune dysfunction. Collectively, these factors led to the perception that cancer vaccines were biologically interesting but clinically impractical.
Core scientific and clinical limitations of earlier approaches
Antigen selection (shared TAAs) and immune tolerance: Most first-generation therapeutic cancer vaccines were built around shared tumour-associated antigens (TAAs), proteins overexpressed in cancer cells but also present at low levels in normal tissues. Because the immune system is trained to tolerate self-antigens, these targets had limited capacity to generate strong cytotoxic T-cell responses. This biological constraint was reflected in several high-profile late-stage failures including GSK’s MAGE-A3 programme (in melanoma and NSCLC), Merck KGaA’s tecemotide (L-BLP25), which targeted the MUC1 glycoprotein in NSCLC, and Bavarian Nordic’s PROSTVAC, a PSA-targeted viral vaccine for prostate cancer.
TME and immune escape: A second major limitation was the inability of early vaccine strategies to overcome the TME. First-generation cancer vaccines were commonly administered as monotherapies in patients with advanced metastatic disease, where tumour burden is high and immunosuppressive signalling is well established. Even when tumour-specific T cells were successfully generated in the periphery, their infiltration and cytotoxic function within the tumour were actively inhibited by a complex network of suppressive signals. This included upregulation of immune checkpoints such as PD-L1, recruitment of regulatory T cells and myeloid-derived suppressor cells (MDSCs) and secretion of inhibitory cytokines such as TGF-β and IL-10.
Disease setting and tumour burden: Clinical positioning was another critical determinant of failure. Early vaccine trials were conducted predominantly in late-stage, heavily pre-treated patients with high tumour burden and often impaired immune function. In this context, the time required to generate an adaptive immune response, typically several weeks to months, is biologically and clinically mismatched with the rapid disease progression typical of advanced cancer. In contrast, it is now widely recognised that cancer vaccines are most effective in the minimal residual disease or adjuvant setting, where tumour burden and subclonal diversity are lower and immune competence is relatively preserved.
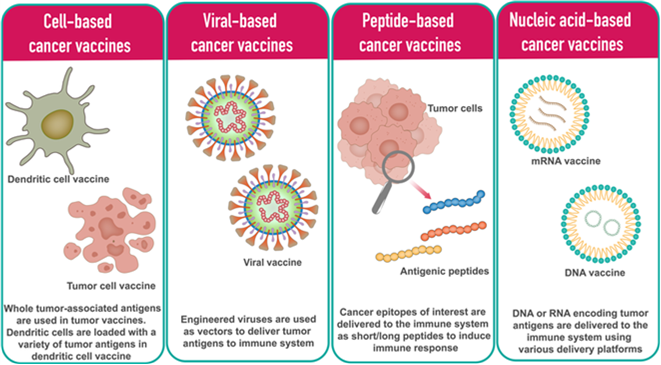
Platform and delivery limitations: Many peptide-based vaccines lacked sufficiently potent or appropriately tailored adjuvants to stimulate dendritic cell maturation and effective cross-presentation through MHC Class I pathways, which is essential for generating CD8+ cytotoxic T-cell responses. Without robust innate immune activation, adaptive responses remained quantitatively and qualitatively inadequate. Modern platforms, including mRNA, viral vectors and next-generation dendritic cell approaches (Exhibit 5) are specifically designed to address this limitation by integrating antigen delivery with strong innate immune stimulation.
Exhibit 5: Different types of therapeutic cancer vaccines
Source: mRNA vaccines for cancer immunotherapy, Yashavantha Vishweshwaraiah, Nikolay Dokholyan. 2022. Frontiers of Immunology
Field at an inflection point
The resurgence of cancer vaccines is not driven by a single breakthrough but by the convergence of multiple enabling technologies.
Neoantigen targeting – moving beyond shared self-antigens: Next-generation cancer vaccines prioritise tumour-specific neoantigens rather than shared TAAs, avoiding central immune tolerance and enabling high-avidity cytotoxic T-cell responses. This shift materially improves the probability of meaningful tumour control, as neoantigens are recognised as non-self and can drive polyclonal immune activation through multi-epitope designs. Clinical validation is emerging across multiple platforms. Moderna/Merck’s V940 has demonstrated a significant reduction in melanoma recurrence in the adjuvant setting in a randomised Phase IIb trial, while BioNTech/Genentech’s autogene cevumeran has generated strong neoantigen-specific T-cell responses in pancreatic cancer. Viral and epitope-based approaches from Nouscom (NOUS-PEV), OSE Immunotherapeutics (Tedopi) and Transgene (TG4050) further underscore the central importance of antigen selection, alongside optimised delivery technologies, in driving immunogenicity and clinical activity.
OSE Immunotherapeutics – Tedopi: Tedopi is a multi-epitope peptide cancer vaccine targeting five tumour-associated antigens in NSCLC. It is being evaluated in the Phase III ARTEMIA trial as a second-line monotherapy in patients who develop resistance after initial response to ICIs, a rapidly expanding population with limited treatment options. If able to re-sensitise tumours to immune control, Tedopi could address a clear post-checkpoint unmet need and benefit from the growing ICI-treated patient pool.
Transgene – TG4050: TG4050 is an individualised neoantigen vaccine based on Transgene’s myvac platform and delivered via an Modified Vaccinia Ankara (MVA) viral vector. Following tumour sequencing, selected patient-specific neoantigens are encoded to induce a potent CD8+ T-cell response. It is currently in the randomised Phase II of a Phase I/II study in resected head and neck cancer, positioning the programme in the adjuvant setting where immune priming and relapse prevention are most biologically relevant.
Genetic delivery platforms – mRNA and DNA as scalable backbones: New-generation cancer vaccines are increasingly using genetic material, such as mRNA or DNA, to instruct the body’s own cells to produce tumour antigens, rather than introducing the antigens directly as proteins or peptides. This approach helps the immune system recognise the targets more effectively and generates both killer (CD8+) and helper (CD4+) T cell responses. It also naturally activates the early immune signals needed for a strong and durable reaction, reducing the need for additional adjuvants. Moderna’s personalised mRNA vaccines and BioNTech’s personalised and shared antigen programmes (including BNT111) highlight the speed and flexibility of this technology, while Scancell’s DNA ImmunoBody platform (iSCIB1+) shows how a potent off-the-shelf option can be combined with checkpoint inhibitors.
Scancell – iSCIB1+: iSCIB1+ is an off-the-shelf DNA cancer vaccine from its ImmunoBody platform designed to induce tumour-specific T-cell responses and enhance checkpoint inhibitor efficacy in advanced melanoma. The programme has successfully completed the 140-patient Phase II SCOPE study in combination with nivolumab (Opdivo) + ipilimumab (Yervoy), demonstrating a strong progression-free survival signal in a defined (human leukocyte antigen (HLA)-selected population. Following this data, the FDA has cleared the investigational new drug application for a global registrational Phase III trial, expected to initiate in 2026, positioning the asset as a late-stage, potential SoC-expanding combination therapy.
Designed for checkpoint inhibitor synergy: Newer cancer vaccines are also being positioned as immune primers for checkpoint blockade rather than as standalone therapies. The vaccine trains the immune system to recognise the tumour and brings T cells into the tumour, while drugs such as PD-1, PD-L1 or CTLA-4 inhibitors stop those T cells from being switched off. Together, this helps overcome two key problems: many tumours do not already have tumour-fighting T cells, and the tumour environment suppresses immune activity. The positive data for V940 with pembrolizumab show this approach can work in practice. Similar combination strategies, including Scancell’s iSCIB1+ with dual checkpoint blockade, also make it easier for vaccines to fit into current treatment pathways.
Earlier disease settings – MRD, adjuvant and maintenance therapy: Cancer vaccine development is moving away from use in very late-stage disease and focusing instead on earlier settings, such as after surgery or when only small amounts of cancer remain (minimal residual disease). At this stage, the immune system is stronger and the tumour burden is low, giving vaccine-activated T cells a better chance to clear remaining cancer cells and provide long-term protection. This also fits better with how vaccines work, as they need time to build an immune response and are less effective in fast-growing metastatic disease. Mendus’s vididencel in acute myeloid leukaemia (AML) after remission and V940 in post-surgery melanoma are good examples. Earlier use also means larger patient populations and longer treatment duration, which improves commercial potential.
Mendus – vididencel: Vididencel is an allogeneic dendritic-cell vaccine in Phase IIb (CADENCE) AML maintenance with SoC oral azacitidine in chemo-fit patients, with a Phase Ib DIVA study in venetoclax + azacitadine treated chemo-unfit patients starting in mid-2026 and a global registrational trial targeted for 2027, alongside a Phase I chronic myeloid leukaemia (CML) programme. Earlier ADVANCE II data showed durable relapse-free and overall survival with a favourable safety profile in MRD-positive patients, supporting the maintenance setting strategy.
Exhibit 6 presents a list of the most clinically advanced cancer vaccine programmes in development.
Exhibit 6: Selection of ongoing clinical-stage cancer vaccine programmes
Deal activity in cancer vaccines is re-accelerating after a prolonged period of limited partnering, reflecting renewed confidence in the modality as clinical data and platform maturity improve. The c $1.25bn BioNTech acquisition of German mRNA vaccine developer CureVac in December 2025 is a clear inflection point. This follows earlier large platform alliances such as BioNTech-Genentech and Moderna-Merck and signals that large pharma now views cancer vaccines as a core long-term pillar of IO. For the broader sector, the transaction raises the strategic value of companies with validated antigen-selection technologies, combination data with checkpoint inhibitors and scalable manufacturing infrastructure, and points to a more competitive partnering and M&A environment.
Cancer vaccines are transitioning from a historically high-risk modality to a combination-driven, platform opportunity, supported by randomised clinical validation in the adjuvant and MRD settings. The shift to neoantigen targeting, genetic delivery and PD-1 synergy materially improves probability of success and commercial scalability. Earlier-line positioning expands patient populations and treatment duration, while recent large-scale transactions signal renewed big pharma appetite. Companies with biomarker-led development, checkpoint-combination data and scalable manufacturing are therefore best placed to secure premium partnering and valuation re-rating.
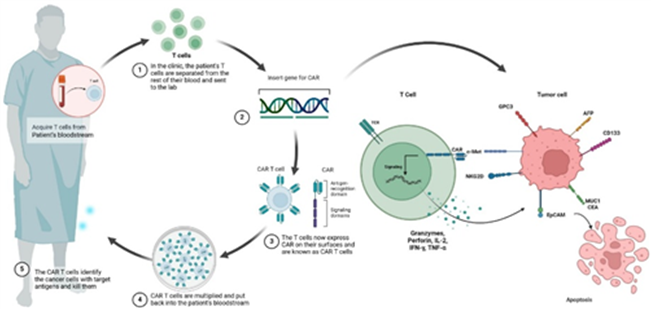
Chimeric antigen receptor-T cell therapies (CAR-Ts) are a form of adoptive cell therapy in which a patient’s immune cells are genetically engineered to recognise and kill cancer cells. The process begins with the collection of T cells from the patient, which are then modified ex vivo to express a synthetic receptor, the chimeric antigen receptor, that binds to a specific tumour surface antigen independently. These engineered cells are expanded and reinfused into the patient, where they proliferate, directly eliminate antigen-expressing tumour cells and can persist as a living drug capable of long-term immune surveillance (Exhibit 7).
Curative potential but concentrated to haematology: To date, CAR-Ts have been applied in the treatment of haematological cancers by targeting the CD19 and BCMA antigens. Approved CAR-T therapies from companies such as Gilead/Kite (Yescarta, Tecartus), BMS (Breyanzi, Abecma) and Johnson & Johnson/Legend (Carvykti) have transformed outcomes in relapsed or refractory B-cell malignancies and multiple myeloma, delivering deep and durable responses in heavily pre-treated patients (Exhibit 8). This success stems from several structural advantages over conventional therapies: high target specificity, in vivo expansion after infusion and the potential for durable remission following a single treatment. However, the treatment is also associated with unique immune-mediated toxicities. Complex autologous manufacturing and the need for specialised centres for treatment delivery currently limit scalability. More importantly, clinical activity in solid tumours has been modest (due to tumour heterogeneity), highlighting that first-generation autologous CAR-T designs are highly effective in liquid cancers but not yet broadly applicable across oncology.
Exhibit 8: FDA-approved CAR-T therapies
Source: Edison Investment Research
Antigen escape – The shift to multi-target constructs: Antigen escape in CAR-T cell therapy is a mechanism of treatment resistance where tumour cells evade destruction by losing or reducing the specific surface protein (antigen) that the CAR-T cells are engineered to recognise. Relapse driven by antigen loss is a major limitation of single-target CAR-Ts. Dual and multi-antigen targeting is now a central development focus, designed to address this limitation by simultaneously recognising more than one tumour antigen, reducing the probability of immune evasion and improving depth and duration of response. Academic and China-based programmes evaluating dual CD19/CD22 CAR-Ts, as well as BCMA/CD19 constructs being explored by multiple developers, exemplify this strategy. In parallel, next-generation single-target approaches such as Autolus’s obe-cel (Aucatzyl), approved by the FDA in November 2024 for adults with relapsed/refractory B-cell precursor ALL, use a fast off-rate CD19 binder to enhance T-cell persistence and reduce exhaustion, representing an alternative strategy to improve durability of response.
This strategy is particularly relevant in B-cell malignancies and multiple myeloma, where antigen downregulation is well documented. These designs are expected to become increasingly important as CAR-Ts move into earlier lines of therapy where durability requirements are higher.
Toxicity and safety – Enabling broader adoption: Cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS) remain key barriers to treatment in community settings
CRS occurs when activated CAR-T cells trigger massive cytokine release, leading to systemic inflammation. Over 70% of patients undergoing treatment with certain CAR T-cell therapies or T-cell engaging bispecific antibodies are affected. While tocilizumab (anti-IL-6) has improved CRS management, prevention remains a critical unmet need.
ICANS affects 20–60% of CAR-T patients, presenting as confusion, aphasia, seizures and, in severe cases, cerebral oedema.
Engineering approaches to reduce tonic signalling and control CAR activation are being pursued by Autolus and Allogene, while pharmacological strategies targeting inflammatory pathways are emerging as a complementary solution.
In parallel, adjunctive therapies specifically targeting CRS are being developed. A notable example is Poolbeg Pharma’s POLB-001, an oral p38 MAP kinase inhibitor, being developed to prevent CRS associated with CAR-T and bispecific antibodies. The asset has completed Phase I human challenge work demonstrating broad cytokine suppression and is now entering a Phase IIa (TOPICAL) proof-of-concept study in r/r multiple myeloma patients receiving the bispecific T-cell engaging antibody teclistamab, with interim data expected in summer 2026. The programme has received FDA Orphan Drug Designation for CRS prevention, positioning it as a potential first-in-class prophylactic that could enable outpatient delivery and broader adoption of cell and T-cell-redirecting therapies.
Manufacturing bottlenecks – the move to allogeneic, off-the-shelf platforms: Off-the-shelf (allogeneic) CAR-T therapies use pre-made immune cells from healthy donors instead of creating a custom product for each patient. This could allow treatment to be given much faster, at lower cost and with more consistent quality. Gene editing removes the donor cell’s natural receptor so it does not attack the patient and is less likely to be rejected, an approach being developed by companies such as Allogene and Cellectis. Newer ‘immune-cloaked’ versions use CRISPR to help the cells stay in the body longer, which is important for long-lasting benefit. Another step forward is induced pluripotent stem cell (iPSC)-derived CAR-Ts, where stem cells are used to produce a large, standardised supply. If these therapies work as well as current CAR-T therapies, they could be delivered more widely and earlier in treatment.
Alternative effector cells: CAR-NK and CAR-iNKT: Other immune cells such as natural killer (NK) cells and invariant natural killer T (iNKT) cells offer several structural merits over conventional CAR-T cells. They are inherently allogeneic, have a lower risk of graft-versus-host disease and appear to produce less severe CRS. Nkarta and Fate Therapeutics are advancing CAR-NK programmes in both haematological and solid tumours, while Arovella Therapeutics is developing CAR-iNKT therapies, with encouraging early data. These platforms may be particularly important for solid tumours and outpatient delivery models. A key current limitation however is the relatively short in-vivo persistence and limited clinical durability particularly for CAR-NK approaches, which may necessitate repeat dosing to match the long-term efficacy achieved with autologous CAR-T therapies.
CAR-T for solid tumours – the largest expansion opportunity: Extending CAR-T therapies into solid tumours represents the biggest long-term growth opportunity for the modality, but also the most complex biologically due to the immunosuppressive TME and heterogeneous antigen expression. Armoured CAR-T cells, designed to secrete stimulatory cytokines such as IL-12 or IL-15, aim to remain active within this hostile environment and recruit other immune cells; this approach is being explored in multiple early-stage clinical and academic–industry programmes. To improve tumour specificity and safety, logic-gated CAR designs that require the presence of more than one tumour antigen for activation are also under development. Modular adaptor CAR platforms, such as Prescient Therapeutics’ OmniCAR, provide an alternative strategy by enabling controllable and retargetable tumour recognition. Most of these approaches remain in early phases of clinical development.
Expanding beyond oncology: CAR-T therapies are emerging as a potentially transformative modality in autoimmune disease by delivering deep and durable depletion of pathogenic B cells, creating the possibility of a one-time immune reset rather than chronic immunosuppression. Early academic CD19 CAR-T studies in systemic lupus erythematosus have demonstrated drug-free remissions, catalysing rapid industry investment. A key requirement for broader adoption in immunology is an improved safety and tolerability profile suitable for outpatient use. Immix Biopharma’s NXC-201, a BCMA-targeted CAR-T in Phase Ib/II for AL amyloidosis with registrational intent, uses a sterically optimised design that has shown low neurotoxicity and short-duration CRS. This profile underpins planned expansion into B-cell-driven non-oncology indications, where risk tolerance is lower but the commercial opportunity is significantly larger.
In-vivo CAR-T: Next-generation cell therapy without ex-vivo manufacturing: An area of development that has been gaining increasing interest is in-vivo CAR-T therapy, an emerging approach in which a patient’s T cells are genetically programmed directly inside the body using targeted delivery technologies such as lipid nanoparticles or viral vectors, removing the need for complex ex-vivo cell processing. This has the potential to significantly reduce manufacturing time, lower cost of goods and enable treatment in outpatient or community settings, addressing key access and scalability barriers associated with current autologous CAR-T therapies. The modality may also allow repeat dosing and more controlled cell expansion, although this remains to be demonstrated clinically. This field has seen increasing M&A and licensing activity in the past year and is becoming one of the most commercially attractive areas of development within the cell therapy space. Exhibit 9 highlights some of the major deals in this space, the most recent being the acquisition of Orna Therapeutics by Eli Lilly for up to $2.4bn in February 2026.
Competitive pressure from bispecific antibodies: Bispecific T-cell engagers from companies such as Amgen (Blincyto), Regeneron and Roche are achieving high response rates in haematological malignancies with an off-the-shelf format, creating both a competitive threat and a complementary treatment option. Their ease of administration and lower upfront cost are accelerating adoption, particularly in earlier lines of therapy. As a result, CAR-T developers are increasingly focused on demonstrating superior durability and curative potential to maintain differentiation.
The long-term value of CAR-T therapy will depend on its evolution from an autologous, late-line therapy into a scalable, earlier-line treatment modality. Off-the-shelf manufacturing, improved safety profiles and expansion into solid tumours are the key inflection points. Companies that can demonstrate durable responses with faster delivery and outpatient administration are likely to capture a disproportionate share of future growth.
Other promising treatment modalities
Emerging intratumoural modalities
Intratumoural delivery is gaining traction as a strategy to activate the anti-tumour immune response directly at the tumour site while minimising systemic toxicity. By converting immune-cold lesions into inflamed, T cell-infiltrated tumours, these approaches can generate systemic immunity and improve responses to checkpoint inhibitors. Rather than competing with established backbones, most intratumoural therapies are being developed as immune-priming combination partners, with their greatest potential in accessible solid tumours and earlier treatment settings where immune function is less compromised.
Oncolytic viruses: Genetically engineered viruses selectively replicate in tumour cells, causing lysis and releasing tumour antigens together with strong inflammatory signals. This drives T-cell priming and can induce responses in distant, non-injected lesions. Amgen’s T-VEC, the first approved agent in this class, established clinical proof of concept in melanoma and continues to be evaluated in PD-1 combinations. Other advanced stage programmes include CG Oncology’s CG0070 and Candel’s CAN-2409, with value centred on checkpoint combinations
Non-viral oncolytic therapies: These agents replicate the in-situ vaccination effect without using a viral vector, allowing simpler manufacturing and repeat dosing. Lytix’s LTX-315 is the lead example, a synthetic oncolytic peptide that disrupts tumour cell membranes and induces rapid immunogenic tumour cell death and systemic CD8+ T-cell responses, with a favourable safety profile for combination strategies.
STING agonists: Intratumoural STING activation drives type-I interferon signalling and dendritic-cell priming at the source of the antigen. Local delivery improves the therapeutic window and provides a strong mechanistic rationale for use with PD-1 inhibitors. Programmes are being advanced by companies including Takeda and BMS.
Toll-like receptor (TLR) agonists: TLR9 agonists such as Regeneron’s vidutolimod (CMP-001) enhance antigen presentation and T-cell infiltration when injected into tumours, with clinical activity in checkpoint-refractory melanoma and head and neck cancer.
Immuno-modulatory antibodies – reprogramming the TME
Immuno-modulatory antibodies are an emerging class of IO agents that work by changing the tumour’s immune environment rather than attacking cancer cells directly. They act on both the innate and adaptive immune system to improve antigen presentation, boost antibody-driven tumour killing and help overcome resistance to PD-1 inhibitors and other targeted antibodies, making them important combination partners. BioInvent’s BI-1206, a first-in-class FcγRIIB-blocking antibody in Phase I/II, is a key example, restoring sensitivity to rituximab in lymphoma with early signs of re-sensitising tumours to pembrolizumab in solid tumours. Overall, these therapies act as immune amplifiers that increase response rates to established checkpoint backbones and are an important source of the next wave of IO value.
Pharma patent cliff – implications
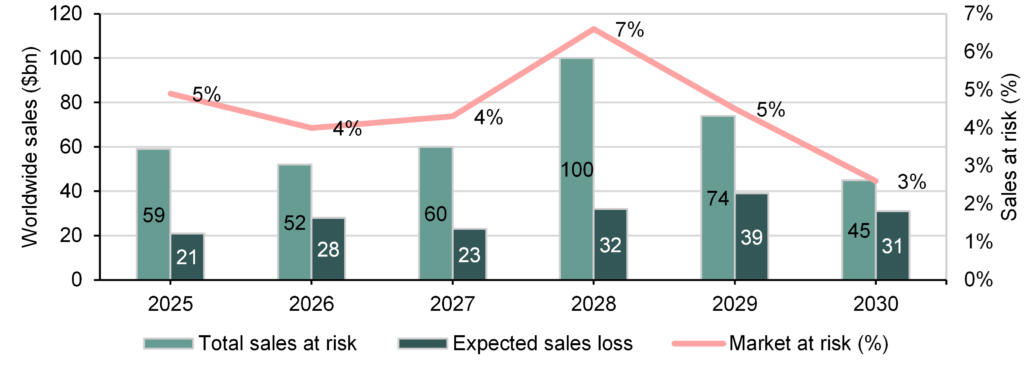
The healthcare sector is approaching an unprecedented loss-of-exclusivity cycle, with >$200bn of branded drug revenue at risk by 2030 (Exhibit 10). The concentration of exposure in a small number of megabrands means the impact will be both financially material and strategically disruptive for large pharma, which will be looking to acquire new assets to replenish pipelines and future revenues.
Exhibit 10: Worldwide sales at risk from patent expiration (2025–30)
The notable event in this cycle is the expected biosimilar exposure of Merck’s Keytruda from 2028, currently the world’s largest oncology drug with c $31.7bn in sales in 2025. BMS’s Opdivo, with over $10bn in revenue in 2025, faces a similar timeline. Together, these two PD-1 inhibitors account for over $40bn of at-risk revenue, making IO one of the largest contributors to the late-decade patent cliff. This is structurally different from previous cycles because PD-1 therapy is not a single indication franchise, but a treatment backbone across multiple tumour types and lines of therapy.
What it means for the biotech sector
Lifecycle management is necessary but not sufficient: Large pharma is already implementing lifecycle strategies to defend these franchises, most visibly through subcutaneous formulations of PD-1 antibodies such as Keytruda and Opdivo. However, lifecycle management alone is unlikely to offset the scale of the revenue decline. The more durable strategy is to build proprietary combination regimens that extend the clinical relevance of the checkpoint backbone, which in turn increases reliance on external innovation.
Combination-driven innovation – a structural tailwind for biotechs: The Merck-Moderna collaboration around the personalised cancer vaccine V940 illustrates how the patent cliff is reshaping deal-making. Positive Phase II melanoma data in combination with Keytruda led to a global profit-share agreement, reflecting the value of assets that increase responder rates and enable earlier-line use. BioNTech’s neoantigen vaccine partnership with Genentech follows the same logic. These therapies do not compete with PD-1; they expand its addressable population, which makes them strategically attractive and allows smaller companies to monetise early clinical proof of concept.
Cell therapy/CAR-T platform acquisitions and modality leadership: A similar dynamic is evident in cell therapy, where the focus has shifted to scalable next-generation platforms. AbbVie’s acquisition of Capstan for up to $2.1bn, AstraZeneca’s purchase of EsoBiotec for up to $1bn and Gilead/Kite’s acquisition of Interius for $350m all target in-vivo cell-engineering technologies that remove the manufacturing bottlenecks of autologous CAR-T therapies. Eli Lilly’s recent acquisition of Orna for up to $2.4bn reinforces the strategic importance of programmable immune-engineering platforms. These transactions show that modality leadership alone can crystallise significant value for small biotechs, often before pivotal data.
Targeting resistance – the rise of immune-modulatory combinations: Overcoming resistance to PD-1 and other targeted therapy is another major investment theme. BioInvent’s BI-1206, which restores sensitivity to rituximab and pembrolizumab by targeting FcγRIIB. CD47/SIRPα programmes, including assets originating from Trillium (Pfizer) and the AbbVie/I-Mab collaboration, aim to enhance macrophage-mediated tumour cell clearance, although recent clinical setbacks across the class have increased the focus on differentiated safety and combination positioning. By increasing response rates rather than competing with established backbones, these therapies extend the commercial lifespan of PD-1 and are therefore highly attractive combination partners.
From single-asset licensing to platform consolidation: The urgency to replace multi-billion-dollar revenues is also changing the structure of deal-making. Large pharma is increasingly acquiring entire technology platforms, as seen in BioNTech’s $1.25bn acquisition of CureVac and Eli Lilly’s acquisition of Orna. Owning the underlying technology allows rapid generation of multiple clinical assets and shortens time to impact. For small biotechs, this creates an opportunity to monetise platform value earlier and reduces dependence on late-stage, capital-intensive development.
Financing and valuation implications for the biotech sector: The patent cliff creates a multi-year period of structural demand for external innovation. Assets that demonstrate clear synergy with PD-1, durable responses, or the ability to move into earlier lines of therapy are likely to attract partnering interest earlier, at Phase I or II, bringing forward value inflection points. At the same time, the bar for differentiation is rising, with increasing emphasis on biomarker selection, durability and health-economic benefit.
Tougher evidence bar and payer friction post-loss of exclusivity: As pricing dynamics intensify around megabrands, payers will push harder on incremental benefit. Biotechs should expect increasing emphasis on: clear biomarkers, durable endpoints, health-economic outcomes (reduced hospital time, fewer cycles) and differentiated safety/route of administration. In the US, the Inflation Reduction Act and Medicare price negotiation for high-spend biologics further increase long-term pricing pressure, reinforcing the need for clinically differentiated, earlier-line and combination-driven strategies to sustain premium reimbursement.
What it means for investors
Lower commercial risk: From an investor standpoint, IO remains one of the most compelling areas in biotechnology because it combines large commercial markets, multiple value-creation inflection points and strong strategic demand from large pharma. Unlike many therapeutic areas where new entrants must displace entrenched standards of care, most next-generation IO assets are largely developed as combination partners to existing treatments. This significantly lowers commercial adoption risk and increases the probability of partnering at an early clinical stage. For investors, this creates a model in which Phase I/II proof-of-concept data can drive disproportionate valuation re-rating, well ahead of registrational timelines.
Pipeline-in-a-product optionality: Many IO modalities are inherently modular and can be expanded across multiple tumour types and treatment settings. Personalised cancer vaccines, cell therapies and immune-modulatory antibody platforms can generate several clinical programmes from a single technology base. This supports higher risk-adjusted valuations, multiple deal opportunities and lifecycle expansion, which is particularly valuable for small and mid-cap companies.
Accelerated development pathways: IO assets often target high-unmet-need or biomarker-selected populations, making them well suited for expedited regulatory pathways and smaller, faster clinical trials. The use of durable endpoints such as long-term survival or MRD clearance can shorten development timelines and reduce funding requirements, improving the return profile for investors.
Premium pricing: Therapies that deliver deep and durable responses, particularly those with curative potential, can command premium pricing despite high upfront costs. This is already evident in the CAR-T space (eg Kymriah and Yescarta come with list price of $47k and $373k, respectively) and is likely to extend to immune-reset approaches in both oncology and autoimmune disease. Strong pricing power translates into attractive peak-sales potential and supports higher valuation multiples.
Structural deal flow driven by the patent cliff: The loss of exclusivity for major ICIs is creating a multi-year need for revenue replacement among large pharmaceutical companies. This is accelerating licensing and M&A activity for assets that can expand the PD-1 ecosystem or overcome resistance. For investors, this provides a clear and recurring exit pathway through strategic transactions.
Multiple value inflection points across the development cycle: IO programmes typically generate several catalysts, including early immune-response readouts, combination data, expansion into new indications and regulatory designations. This steady flow of milestones supports sustained investor engagement and allows value to be realised progressively rather than only at approval. From a capital markets perspective, IO offers earlier value inflection than most therapeutic areas. Immune-response readouts, durability signals and initial combination data can drive meaningful re-rating well ahead of approval, while sustained M&A demand provides a clear monetisation pathway.
Risk-reward considerations
Investment risks
Despite its transformational clinical impact and strong deal activity, immuno-oncology investments continue to carry meaningful risk.
Clinical development risks:
Biology remains complex: Response rates to checkpoint inhibitors remain limited in many tumour types, and overcoming primary and acquired resistance is still a major scientific challenge.
Translation risk in novel modalities: Next-generation approaches such as cancer vaccines, in-vivo cell therapy and myeloid targets have a strong mechanistic rationale but limited late-stage validation.
Combination dependency: Most emerging IO therapies are developed on top of PD-1 backbones, making clinical success dependent on demonstrating incremental benefit over an already effective SoC.
Safety and tolerability: Immune-related adverse events, CRS in cell therapies and overlapping toxicities in combinations can limit dose intensity and adoption.
Commercial risks:
PD-1 market saturation: The SoC is well established across multiple tumour types, raising the efficacy bar for new entrants and increasing the need for clear differentiation.
Competitive intensity: Many tumour settings are crowded with similar mechanisms, increasing the risk of being clinically or commercially marginalised.
Pricing and payer scrutiny: As combination use expands, payers are increasingly focused on cost-effectiveness and duration of therapy, particularly in large solid-tumour indications.
Operational complexity for advanced therapies: Cell therapies and personalised vaccines require specialised manufacturing, logistics and treatment centres, which can limit scalability.
Company-specific risks:
Single-asset exposure: Many small IO companies are built around one lead programme, making clinical readouts binary valuation events.
Funding requirements for combination trials: Large, randomised studies in earlier lines of therapy are capital intensive and often beyond the reach of smaller balance sheets without a partner.
Platform execution risk: Failure of the lead asset can have a negative impact on confidence in the broader technology platform.
Mitigating factors
Several factors help mitigate traditional IO investment risks.
Strong external demand for innovation
Patent-cliff driven deal flow: Loss of exclusivity for PD-1 franchises is creating a structural need for revenue replacement, increasing partnering probability for differentiated assets.
Combination relevance lowers commercial risk: Therapies that enhance checkpoint activity can integrate into existing treatment paradigms rather than displace them.
Increasingly validated biology
Clinical PoC for new approaches: Recent positive data in areas such as personalised cancer vaccines and next-generation cell therapies validate previously unproven mechanisms.
Biomarker-driven development: Improved patient selection increases the probability of trial success and supports premium pricing.
Better understanding of resistance pathways: Growing insight into the TME enables more rational combination strategies.
Large and durable market opportunity
Oncology remains the largest therapy area globally: IO continues to capture a disproportionate share of oncology drug spending.
Earlier-line treatment expansion: Movement into adjuvant and MRD settings increases addressable patient populations and treatment duration.
Potential beyond oncology: Immune-reset approaches in autoimmune disease represent a significant long-term optionality for several IO modalities.
Platform and lifecycle optionality
Pipeline-in-a-product dynamics: Single technologies can be applied across multiple tumour types and combinations, creating repeated value inflection points.
Multiple routes to monetisation: Licensing, co-development and M&A provide exit opportunities well before commercialisation.
Regulatory acceleration: Breakthrough, Fast Track and orphan designations can shorten timelines, increase market exclusivity and reduce capital requirements.
Investment conclusions
The IO landscape represents one of the most compelling long-term investment themes in healthcare today, driven by scientific validation, platform technologies and the need to replace multibillion-dollar checkpoint revenues. The shift from monotherapy checkpoints to combination-based, immune-ecosystem control marks the next phase of value creation.
Several factors make the current investment environment particularly attractive. The upcoming PD-1 patent cliff is accelerating external innovation and increasing partnering appetite for assets that expand responder populations or overcome resistance. At the same time, advances in biomarker-driven development, cell-engineering, mRNA and in-situ tumour-priming approaches are improving clinical success probabilities and shortening development timelines.
The commercial opportunity remains substantial. Oncology continues to be the largest therapeutic area globally, with immuno-oncology capturing a disproportionate share of drug spending. Movement into earlier lines of therapy, adjuvant and MRD settings is expanding the addressable population, while durable responses and treatment-free remission support premium pricing and strong pharmacoeconomic value.
For investors, immuno-oncology offers multiple avenues for exposure. Large-cap pharmaceutical companies provide franchise stability and lifecycle management of checkpoint backbones, mid-cap innovators offer focused exposure to next-generation modalities and small-cap platform companies present the most asymmetric risk-reward, particularly where early data can unlock partnering or acquisition.
The companies highlighted in this report reflect the breadth of innovation across the field. From personalised cancer vaccines and next-generation cell therapies to immune-modulatory antibodies and intratumoural immune-priming approaches, these platforms are designed to extend the PD-1 ecosystem rather than compete with it, which lowers commercial adoption risk.
Risk mitigation comes from the structural demand for external innovation and the modular nature of many IO technologies. Multiple value inflection points, accelerated regulatory pathways and the ability to monetise assets at Phase I/II through partnerships help offset traditional biotech funding and development risks.
As this next phase of immuno-oncology unfolds, investors who position themselves early in differentiated platforms and combination-relevant assets are likely to benefit from sustained deal flow and valuation re-rating. The sector combines a large market opportunity, strong strategic buyer demand and scalable technologies, creating a highly attractive long-term investment landscape.
Appendix: Company showcase
The following companies represent a selection of the most innovative approaches across the immuno-oncology landscape, each targeting a key biological or commercial bottleneck such as responder expansion, resistance reversal, immune priming or scalable cell therapy delivery (Edison clients are indicated with an asterisk).
BioInvent International
Advancing immuno-oncology through precision antibody therapeutics
BioInvent International AB (Nasdaq Stockholm: BINV) is a clinical-stage biotech focused on developing first-in-class immune-modulatory antibodies to address key limitations in cancer immunotherapy, particularly treatment resistance and low response rates. Its antibodies target immune checkpoints and tumour microenvironment biology to unlock new mechanisms of action.
The company’s discovery engine is built on its proprietary F.I.R.S.T platform, which enables the simultaneous identification of novel targets and corresponding antibodies directly from patient-derived tumour material. This is complemented by the n-CoDe antibody library, supporting rapid generation of differentiated candidates.
Following a strategic portfolio prioritisation in August 2025, BioInvent is focusing resources on its two lead assets, BI-1206 and BI-1808. BI-1206 (anti-FcγRIIB) is designed to restore sensitivity to anti-CD20 therapies in non-Hodgkin’s lymphoma and enhance responses to PD-1 inhibitors in solid tumours. In a Phase I/IIa trial with rituximab and acalabrutinib, early data from the triple-combination arm showed 100% disease control and a 63% objective response rate. Subsequent data for the Phase IIa safety run-in (n=15) showed an overall response rate of 80%, with 47% achieving complete responses. The disease control rate was 100%.
A second Phase IIa study combining BI-1206 with pembrolizumab in first-line NSCLC and uveal melanoma marks its entry into treatment-naïve settings. The asset has also been reformulated for subcutaneous delivery to improve convenience, safety and durability.
BI-1808 targets TNFR2 to counter tumour immune evasion by depleting regulatory T cells and expanding CD8+ T cells. In Phase IIa cutaneous T-cell lymphoma, it has demonstrated strong immune engagement with 100% disease control and a 45% response rate, alongside ongoing evaluation in combination with pembrolizumab in solid tumours.
Both programmes have received supportive FDA designations, including Orphan Drug and Fast Track status, highlighting their potential in high-unmet-need indications. BioInvent’s integrated GMP manufacturing provides scalable, cost-efficient biologics production and ensures reliable global clinical supply, strengthening its operational and partnering position.
Immix Biopharma*
Programming deep and durable responses in AL amyloidosis
Immix Biopharma (NASDAQ: IMMX) is a clinical-stage company focused on advancing next-generation CAR-T therapies for patients with relapsed/refractory AL amyloidosis, a rare plasma cell disorder characterised by rapid organ deterioration and poor outcomes with conventional treatments. Its lead and wholly owned programme, NXC-201, is a BCMA-directed autologous CAR-T therapy designed with the objective of inducing deep haematologic responses and enabling meaningful and potentially durable organ recovery.
Clinical data generated to date highlight compelling efficacy in a heavily pretreated patient population, including high overall and complete response rates and early signals of organ improvement. Recent interim data (20 patients) presented from the ongoing Phase Ib/II NEXICART-2 trial (n=40) showed a 75% complete response (CR) rate, with 15/20 patients achieving deep haematological responses. Of the five patients without a CR, four were MRD negative. Importantly, NXC-201 has demonstrated a favourable safety and tolerability profile, with low rates of high-grade CRS and neurotoxicity, a critical consideration in AL amyloidosis patients who are often frail and ineligible for intensive therapies. This safety profile, combined with rapid manufacturing turnaround and reliable product delivery, supports the potential use of NXC-201 in a broader treatment setting and in patients with significant organ involvement.
The ongoing US-based NEXICART-2 study is intended to support a registrational pathway for AL amyloidosis with a biologics licence application planned before end-FY26. The company was recently granted the Breakthrough Therapy Designation for NXC-201 by the FDA based on the strength of the interim data. NXC-201 also holds the Regenerative Medicine Advanced Therapy designation by the US FDA and Orphan Drug Designation by FDA and the EMA.
With CAR-T increasingly recognised as a transformative modality in non-oncology indications, NXC-201 is positioned to address a critical gap for AL amyloidosis patients who have limited therapeutic options and poor prognosis following relapse.
Immutep
Targeting LAG-3 Biology Across Cancer and Autoimmunity
Immutep (ASX: IMM) is a late-stage biotechnology company and a pioneer in therapies targeting the MHC Class II and LAG-3 pathways for cancer and autoimmune diseases. Its scientific leadership is rooted in the discovery of LAG-3 by CSO Dr Frédéric Triebel, positioning the company at the forefront of this immune checkpoint field. Immutep’s approach focuses on modulating key regulators of the immune response to enhance anti-tumour activity and improve outcomes for patients who derive limited benefit from existing immunotherapies.
The oncology pipeline is anchored by two clinical-stage assets that employ complementary strategies to stimulate anti-cancer immunity. Efti (eftilagimod alpha) is a first-in-class MHC Class II agonist that directly activates antigen-presenting cells (APCs), including dendritic cells and monocytes. By engaging this central immune control point, efti drives a broad and durable immune response, priming CD8+ cytotoxic T cells and expanding multiple immune effector populations. This mechanism has demonstrated strong synergy with anti-PD-(L)1 therapy, chemotherapy and radiotherapy, alongside a favourable safety profile across combinations.
Efti is currently being evaluated in TACTI-004 (KEYNOTE-F91), a pivotal Phase III trial in partnership with MSD (n=750), in combination with pembrolizumab and chemotherapy as first-line treatment for advanced/metastatic NSCLC (50% enrolment completed). The study builds on positive data from TACTI-002 and INSIGHT-003 (>165 patients), which showed high response rates and encouraging survival outcomes. Notably, efficacy has been observed across the full PD-L1 expression spectrum, including the large low/negative PD-L1 population with significant unmet need. Efti is also being explored in multiple additional solid tumours, including head and neck, urothelial, breast cancer and soft tissue sarcoma.
Immutep’s second clinical asset, LAG525 (ieramilimab), the first anti-LAG-3 antibody, is out-licensed to Novartis, validating the company’s leadership in LAG-3 biology. Earlier-stage programmes include a small-molecule LAG-3 inhibitor and IMP761, the first LAG-3 agonist antibody for autoimmune disease, which has generated promising initial Phase I data (single ascending dose part completed).
Lytix Biopharma
Developing a new class of intratumoral immunotherapies
Lytix Biopharma (Euronext Growth Oslo: LYTIX) is developing a novel class of oncolytic immunotherapies designed to harness the full potential of the immune system by directly reprogramming the tumour microenvironment. Its lead asset, ruxotemitide (LTX-315), is a first-in-class intratumoral therapy that rapidly disrupts tumour cell membranes, releasing a broad repertoire of tumour antigens and danger signals that drive immune activation. By initiating immune recognition rather than relying on pre-existing anti-tumour immunity, the approach is positioned to convert immunologically ‘cold’ tumours into ‘hot’ tumours and expand the benefit of checkpoint inhibitors beyond current responder populations.
This mechanism enables broad T-cell priming and durable systemic immune responses, providing a strong biological rationale for combination with PD-1/PD-L1 inhibitors in indications where response rates to checkpoint blockade alone remain limited. Clinically, the company is prioritising injectable and resectable solid tumours, where intratumoral delivery is both feasible and therapeutically meaningful.
In partnership with Verrica Pharmaceuticals, ruxotemitide has completed a Phase II study in non-metastatic skin cancer, demonstrating a 97% objective response rate, 51% pathological complete responses and an 86% mean tumour reduction across treated lesions, alongside a favourable safety profile with no serious adverse events. Based on these data and FDA alignment on trial design, Verrica is preparing a pivotal Phase III programme in basal cell carcinoma. Lytix retains rights in metastatic and neoadjuvant settings, including melanoma, triple-negative breast cancer and Merkel cell carcinoma, where combination strategies with checkpoint inhibitors represent a key value driver.
The pipeline also includes LTX-401, an oncolytic candidate targeting liver metastases, extending the platform to deep-seated tumours. With a validated mechanism, encouraging clinical activity and a flexible partnering strategy, Lytix is positioning intratumoral immunotherapy as a complementary modality within the immuno-oncology landscape, aimed at delivering a durable systemic benefit for patients underserved by current treatments.
Mendus*
Programming durable remissions in AML and CML
Mendus (OMX: IMMU) is a clinical-stage immuno-oncology company developing off-the-shelf cellular cancer vaccines designed to induce durable, tumour-specific immune responses and reduce relapse in haematological malignancies and solid tumours. Its lead programme, vididencel, is an allogeneic dendritic cell-based therapy aimed at establishing long-term immune surveillance in patients with AML and CML.
By priming and expanding tumour-specific T cells, vididencel is intended to eliminate residual malignant cells and deliver sustained disease control without the toxicity associated with intensive consolidation treatments. The off-the-shelf format enables rapid treatment, scalable manufacturing and favourable logistics compared with autologous cell therapies.
Clinical data to date from the ADVANCE II trial has demonstrated encouraging immune responses, a favourable safety profile and signals of relapse reduction in AML patients following first line intensive induction chemotherapy. The programme is now being repositioned to address a broader AML population, including ‘chemo-unfit’ patients receiving venetoclax-based regimens, with new studies (CADENCE and DIVA) expected to inform the design of a potential global registrational trial.
Mendus is also expanding into CML, where vididencel is being developed as a potential treatment-free remission, enabling therapy for patients on long-term TKI therapy, representing a large and well-defined commercial opportunity. Vididencel is also being explored in ovarian cancer and has potential applicability in additional tumour settings where immune priming in the minimal residual disease setting could improve long-term outcomes.
Manufacturing is supported through a dedicated GMP partnership with NorthX Biologics, providing scalable, commercial-ready production capabilities and strengthening the pathway to late-stage development and potential out-licensing. The company is funded into 2027 and is seeking partnerships to support late-stage development and maximise the platform’s potential.
See below for our most recently conducted Edison TV executive interview with Mendus.
Exhibit 11: Executive interview with Erik Manting, CEO of Mendus, and Tariq Mughal, CMO
Source: Edison Investment Research
OSE Immunotherapeutics*
Precision Immunotherapy for the ICI-resistant population
OSE Immunotherapeutics (PAR: OSE) is a clinical-stage biotechnology company developing first-in-class immunotherapies across oncology and immuno-inflammation, with a strategy centred on biomarker-driven precision medicine and differentiated T-cell modulation. Its lead oncology programme, Tedopi, is an off-the-shelf, multi-epitope therapeutic cancer vaccine designed to induce tumour-specific cytotoxic T-cell responses in HLA-A2–positive patients and address resistance to ICIs.
Tedopi is being advanced in NSCLC after ICI failure, a setting with high unmet need and limited treatment options. The vaccine targets five tumour-associated antigens and is engineered to generate durable CD8+ T-cell immunity with a favourable tolerability profile compared with chemotherapy. In the Phase III ATALANTE-1 study, Tedopi demonstrated a clinically meaningful survival benefit versus standard chemotherapy in the ICI-resistant population, establishing proof of concept and supporting its ongoing registrational strategy. The confirmatory ARTEMIA Phase III trial, comparing Tedopi with docetaxel, represents the final step toward potential approval, with a futility analysis expected in 2026.
Beyond oncology, OSE’s immunology pipeline provides additional value creation and partnering optionality. The most advanced asset, lusvertikimab (anti-IL-7R), targets a central pathway in T-cell expansion and is being developed in chronic inflammatory diseases (ulcerative colitis, chronic pouchitis and hidradenitis suppurativa) offering a complementary and potentially less capital-intensive route to late-stage development through partnerships.
See below for our most recently conducted Edison TV executive interview with OSE Immunotherapeutics.
Exhibit 12: Executive interview with Silvia Comis MD, chief clinical and medical research officer at OSE Immunotherapeutics
Source: Edison Investment Research
Percheron Therapeutics*
Unlocking the VISTA checkpoint opportunity
Percheron Therapeutics (ASX: PER) is a clinical-stage immuno-oncology company developing next-generation checkpoint inhibitors to address resistance to PD-1/PD-L1 therapies. Its lead asset, HMBD-002, targets VISTA (V-domain Ig suppressor of T-cell activation), an emerging immune checkpoint strongly associated with poor response to existing immunotherapies and with tumours that remain largely unresponsive to current treatment, including pancreatic, ovarian and prostate cancers. By expanding the number of patients who can benefit from checkpoint blockade, VISTA inhibition represents a key opportunity in the evolution of the IO landscape.
HMBD-002 is differentiated by its IgG4 backbone, the same isotype used in pembrolizumab and nivolumab, which is designed to minimise unwanted immune activation and improve tolerability, an important advantage in a class where earlier candidates have been limited by toxicity. Preclinical studies have demonstrated broad anti-tumour activity both as monotherapy and in combination with PD-1 inhibitors, including in triple-negative breast cancer models (UT Southwestern) and in radiotherapy combinations in head and neck cancer (Stanford), supporting a strong biological rationale for use in CPI-resistant disease.
The completed US Phase I study (n=48) showed an excellent safety profile across doses from 20–1,400mg, with no maximum tolerated dose reached due to the low incidence of dose-limiting toxicities. Early signs of clinical activity included tumour shrinkage and durable stable disease in heavily pre-treated patients, supporting advancement into later-stage trials. A modular Phase II study is expected to commence within 2026.
See below for our most recently conducted Edison TV executive interview with Percheron Therapeutics.
Exhibit 13: Executive interview with Percheron Therapeutics CEO, Dr James Garner
Source: Edison Investment Research
Poolbeg Pharma
Solving the CRS bottleneck in T-cell engagers
Poolbeg Pharma (LSE: POLB) is developing POLB 001, a first-in-class, orally administered p38 MAPK inhibitor designed to prevent cancer immunotherapy-induced CRS, one of the most serious and resource-intensive toxicities associated with T-cell-engaging bispecific antibodies and CAR-T therapies. CRS affects more than 70% of patients receiving certain T-cell-redirecting treatments and currently necessitates administration in specialised centres, leading to prolonged hospitalisation, high healthcare utilisation and constrained patient access. With no approved preventative therapies, POLB 001 is positioned to address a critical bottleneck in the delivery and scalability of next-generation immuno-oncology treatments.
By selectively modulating excessive inflammatory signalling without causing broad immunosuppression, POLB 001 aims to improve the safety profile of T-cell-engaging therapies while preserving anti-tumour immune function, an important consideration in heavily pre-treated cancer populations. Enabling safer outpatient or community-hospital administration could significantly expand treatment capacity, improve patient quality of life and unlock additional commercial value across the rapidly growing cell therapy and bispecific antibody markets.
The initial commercial opportunity, focused on multiple myeloma and diffuse large B-cell lymphoma, is estimated to exceed US$10bn, with further upside as T-cell-engaging therapies move into earlier lines of treatment and into solid tumours. The programme has demonstrated a favourable safety and pharmacodynamic profile in preclinical studies and in a human lipopolysaccharide (LPS) challenge model, providing important translational validation ahead of patient studies.
The Phase IIa open-label, single-arm trial is in advanced preparation and will evaluate POLB 001 in approximately 30 relapsed/refractory multiple myeloma patients receiving an approved bispecific antibody (teclistamab), with interim data expected in summer 2026. The study is being led by The Christie NHS Foundation Trust and the Accelerating Clinical Trials network. FDA Orphan Drug Designation provides additional regulatory validation and commercial optionality. On positive data, Poolbeg expects to pursue a partnering strategy, positioning POLB 001 as a potential enabling therapy for the broader immuno-oncology ecosystem.
Scancell
Developing potent and safe active immunotherapies for a cancer-free future
Scancell (LSE: SCLP) is a clinical-stage immuno-oncology company developing targeted active immunotherapies designed to generate durable, tumour-specific T-cell responses and address the large proportion of patients who fail to benefit from checkpoint inhibitors (ICIs). With approximately half of patients not responding to ICIs and five-year survival in stage IV melanoma below 23%, Scancell’s off-the-shelf platforms aim to enhance efficacy without adding toxicity while improving accessibility versus personalised approaches.
The lead asset, iSCIB1+ (DNA Immunobody), is a needle-free DNA vaccine that induces high-avidity T cells against gp100 and TRP-2 melanoma antigens, selected from patients with spontaneous tumour regression. In the Phase II SCOPE study (n=140), iSCIB1+ in combination with ICIs has demonstrated clinically meaningful improvements over historical CPI outcomes. Progression-free survival in the target HLA population reached 78% at 11 months versus ~46% for nivolumab/ipilimumab, with 69% PFS at 22 months across cohorts. Updated data remained encouraging with PFS of 74% at 16 months in the target population. These data support the potential to establish a new standard of care in advanced melanoma. With FDA IND clearance in place, development is being accelerated towards a randomised registrational study in 2026, likely incorporating a precision biomarker. A commercial-scale GMP manufacturing process with strong stability has already been established, supporting future scalability.
The second clinical programme, Modi-1 (Moditope), is a peptide-based active immunotherapy targeting post-translationally modified tumour antigens and is currently in Phase II evaluation in hard-to-treat solid tumours, including head and neck cancer, with further data expected in early 2026.
Beyond vaccines, Scancell’s GlyMab platform, developed within its Glymab Therapeutics subsidiary, generates high-affinity antibodies against tumour-specific glycans, with two assets already licensed to Genmab, providing external validation and potential non-dilutive value.
See below for our recent Edison TV executive interview with Scancell.
Exhibit 14: Executive Interview with Scancell Holdings’ chief medical officer, Dr Nermeen Varawalla
Source: Edison Investment Research
Transgene
Unlocking the power of individualised cancer immunotherapy with TG4050
Transgene (FRA: TNG) is a clinical-stage biotechnology company developing virus-based immunotherapies, with a strategic focus on individualised neoantigen therapeutic vaccines (INTVs) to prevent cancer relapse in early-stage disease. Its lead programme, TG4050, is based on the proprietary myvac platform and is currently being evaluated in the randomised Phase II part of a Phase I/II trial in operable head and neck cancer (HNSCC).
The myvac platform integrates viral vector engineering, artificial intelligence and translational data to deliver precision immunotherapy at clinically relevant timelines. Using a Modified Vaccinia Ankara (MVA) vector, TG4050 encodes patient-specific neoantigens identified through AI-driven algorithms developed with NEC. This approach is designed to generate potent and durable CD8+ T-cell responses against tumour mutations unique to each patient, with the aim of establishing long-term immune surveillance following surgery and standard-of-care treatment. A digitalised, in-house GMP manufacturing process supports scalable production within a timeframe compatible with routine clinical practice.
Clinical data presented at ASCO 2025 demonstrated the feasibility, safety and strong immunogenicity of TG4050, with 100% disease-free survival at a median 30-month follow-up (n=32). Neoantigen-specific CD8+ T-cell responses persisted for more than two years and showed effector and memory phenotypes consistent with durable protection against relapse. Additional translational analyses presented at SITC 2025 confirmed the induction of polyclonal cytotoxic T cells, expansion of both de novo and pre-existing tumour-reactive clones and the development of tissue-resident immune populations, highlighting the potential to overcome resistance in immunologically ‘cold’ tumours.
With proof of concept established, Transgene is advancing TG4050 towards a registrational pathway and exploring expansion into additional early-stage solid tumours. More broadly, myvac represents a scalable platform for a pipeline of individualised vaccines, positioning the company at the forefront of next-generation precision immuno-oncology.
This website uses cookies so that we can provide you with the best user experience possible. Cookie information is stored in your browser and performs functions such as recognising you when you return to our website and helping us understand which section of the website you find more interesting and useful. See our Cookie Policy for more information.
Strictly necessary and functional
These cookies are used to deliver our website and content. Strictly necessary cookies relate to our hosting environment, and functional cookies are used to facilitate social logins, social sharing and rich-media content embeds.
Advertising
Advertising Cookies collect information about your browsing habits such as the pages you visit and links you follow. These audience insights are used to make our website more relevant.
Please enable Strictly Necessary Cookies first so that we can save your preferences!
Performance
Performance Cookies collect anonymous information designed to help us improve the site and respond to the needs of our audiences. We use this information to make our site faster, more relevant and improve the navigation for all users.
Please enable Strictly Necessary Cookies first so that we can save your preferences!